



Thesis below highlights GALT, where we are fundamentally negative on the upcoming Q424 catalyst. Please reach out to @goderguy on Twitter if you think we are wrong in any way!
Setup: We believe the YE’24 phase III readout testing Belapectin in MASH cirrhosis without varices will fail. We justify our short-thesis through five points - laid out below.
Both preclinical & clinical experimental evidence suggest belapectin’s mechanism is not driving the intended downstream effect of Gal-3 inhibition: (a) suppressing myofibroblast creation and (b) reducing fibrosis.
Inappropriate statistical methods: Rerunning analysis under sound statistical principles for the unplanned post-hoc sub-group (basis for phase III trial) reveals a lack of significance once reassessed.
Belapectin’s binding profile to the Gal-3 protein - as characterized by management - creates an open question on binding profile, given the lack of disclosures.
Limited patent life until 2032-2035 (incongruent management commentary; patent flagged as 2032, management flagging up to 2035) removes the likelihood for a favorable acquisition, with a ~2028-2029 phase III completion & approval at the earliest.
High risk of upcoming dilution, compounded by the factors flagged above, will limit stock price appreciation.
(1) Downstream Mechanism: Preclinical & Clinical data shared do not support downstream fibrosis and smooth muscle actin reduction, post addition of belapectin.
Fibrosis
In GALT’s cirrhosis mice experiments, belapectin was shown to reduce both fibrosis and cirrhosis. Another GALT study found that treatment with belapectin improved liver histology in mice with reduction in NASH activity and collagen deposition.
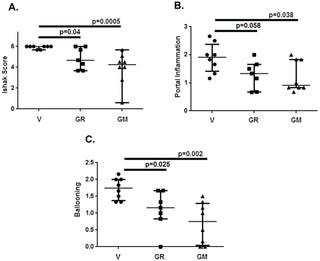
Looking deeper at GALT’s cirrhosis study in mice, we see a reduction in fibrosis vs. vehicle from drug administration (Groups 2-4 represent belapectin in Figure below, Group 1 is the vehicle).
Fibrosis tissue data (Sirius red positive) in groups 1 (vehicle), 2-4 (belapectin), and 5-7 (different drug same mechanism). Groups 2-4 are showing significant reductions in fibrosis (pre-clinical) vs. vehicle (group 1).

However, as seen in panel B in the figure below, the difference in portal inflammation is barely significant (p = 0.038-0.058). Note - the phase III trial primary endpoint is specifically testing for esophageal varices development, which is influenced heavily by portal pressure.
Liver histology data from V (vehicle), GR (belapectin), and GM (different drug similar mechanism)
Therefore, GALT is relying on barely significant data from poorly translatable mouse models, with an unproven binding mechanism (more below), to succeed in this upcoming readout.
Combined with a lack of downstream mechanistic confirmation (smooth muscle actin reduction as flagged below) - we would argue this is a low PoS readout.
Smooth muscle actin reduction
Reductions in smooth muscle actin is an expected downstream impact of Gal-3 inhibition, given reduction in TGF-beta activation, and thus reductions in myofibroblast production which leads to truncated expression of smooth muscle actin.
The authors performed a Western Blot (pre-clinical data) evaluating the differences in expression of smooth muscle actin with belapectin (Groups 2-4). At best, there is only a trend towards reduction in animals treated with belapectin, but the effect was not statistically significant. We have an additional confirmation in the lack of smooth muscle actin inhibition from previous clinical data published by the team.
A lack of clear inhibition of smooth muscle actin - which contributes to the contractility of myofibroblasts and thus influences portal hypertension - likely portends a reduced development cadence for varices (primary endpoint in the phase III trial).
A lack of smooth muscle inhibition calls into question whether myofibroblast production is indeed reduced post belapectin administration. This would either be a result of (1) belapectin NOT efficiently inhibiting Gal-3, and thus not influencing the number of downstream myofibroblasts impacted, or (2) compensatory mechanisms increasing activation of smooth muscle actin, overcoming the inhibition of TGF-Beta signaling (whcih drives a reduction in myofibroblast levels).

The lack of downstream smooth-muscle actin inhibition we see from belapectin administration pre-clinically was confirmed in the clinical phase II trial of belapectin. Similar to the pre-clinical data, this calls into question the mechanism of belapectin, and whether it’s showing a real, downstream effect with inhibition of myofibroblast production. This is important because the main phase III endpoint - reduction in varices, requires a reduction in portal hypertension.
See the third row below in GALT’s phase 2b - the smooth muscle actin staining % was not statistically significant between either experimental group and placebo, which replicates the negative preclinical results for an important mechanism through which their drug could work (note raw values shared below / flagged in study this was not significant).

2) Statistics & Trial Design: Management is putting forward an unplanned post-hoc analysis to drive the basis for a phase III program. We re-run the analysis with a statistically appropriate test (Fisher’s Test over the Chi^2 reported by management), and we adjust for multiplicity to correct for Type I errors, given this was an unplanned sub-group. Resulting p-value is not significant at the required 0.025 level (post Bonferroni correction), and calls into question whether the data used to justify the phase III was actually statistically significant. Additionally - a lack of central reading in the phase II is a confounding difference vs. central readings in a phase III.
In the prior Phase 2b study, GALT administered patients with MASH cirrhosis and portal hypertension belapectin 2 mg/kg (n = 54), 8 mg/kg (n = 54), or placebo (n = 54). The primary endpoint was a change in HVPG at the end of the 52 week period.
The authors found no significant difference in HVPG between the 2 mg/kg or 8 mg/kg belapectin groups and placebo. Belapectin also had no significant effect on fibrosis or NAFLD activity score.
It was only through a post-hoc subgroup analysis of patients without varices at baseline (n = 25 for 2 mg/kg, n = 23 for 8 mg/kg, n = 33 for placebo), that the authors found a significant reduction in HVPG versus baseline. But the effect was only seen in the 2 mg/kg group (p = 0.02) and not the 8 mg/kg subgroup (p = 0.44).
There are a few issues with this analysis:
Management is not adjusting the subgroup - which was not pre-planned - for multiplicity. An appropriate Bonferonni correction would require an alpha threshold of p=0.025 (vs. p=0.05 used), assuming there was only one sub-group analyzed in addition to the active treatment group. If anything - it’s likely management ran multiple post-hoc analyses to find significance, and the appropriate Bonferonni-corrected p-value needs to reduce further beyond 0.025; we are staying conservative here to get to this result.
Lack of dose response is troubling - an effect seen in 2mg/kg and NOT in 8mg/kg for the post-hoc subgroup (isolated to patients without varices upon enrollment) is questionable given the mechanism of belapectin relies on Gal-3 inhibition, whose effect should directly scale with more drug and increased levels of inhibition.
Management’s argument for not seeing a dose response is two-fold:
GCS-100, another galectin-3 inhibitor, saw the same response dynamics in chronic renal disease, where a lower dose of Gal-3 inhibitors showed an effect, vs. a non-effect seen in larger doses of Gal-3 inhibitors.
La Jolla Pharmaceuticals - owners of GCS-100 - argued this was caused by off-target effects on other galectins like galectin-9 overcoming inhibition of Gal-3 alone in high doses. The justification shared here is the fact that complete Gal-3 inhibition was seen at higher doses, and the remaining GCS-100 inhibitors increased the inhibition of Gal-9, which they believe is a pro-fibrotic signal that outweighs the corresponding reduction in Gal-3 inhibition-driven reductions in fibrosis.
Digging deeper - we know that Gal-9 as a molecule exhibits a similar, pro-fibrotic effect to Gal-3. Thus - inhibition of Gal-9 in excess of Gal-3 should only help to reduce fibrosis further - thus the team’s logic is not sound (additional Gal-9 inhibition at a higher dose would be expected to further reduce levels of fibrosis based on the mechanism, vs. increasing the fibrosis levels seen in the trial).
Management is also claiming their drug has a narrow therapeutic window. They found that patients were most likely to respond when the AUC’s were between 3,000 and 12,000 (chart below). If true - per management - they are using this AUC curve to support their hypothesis that the 8 mg/kg group (AUC ~12,000) at the higher end leads to a lack of response.
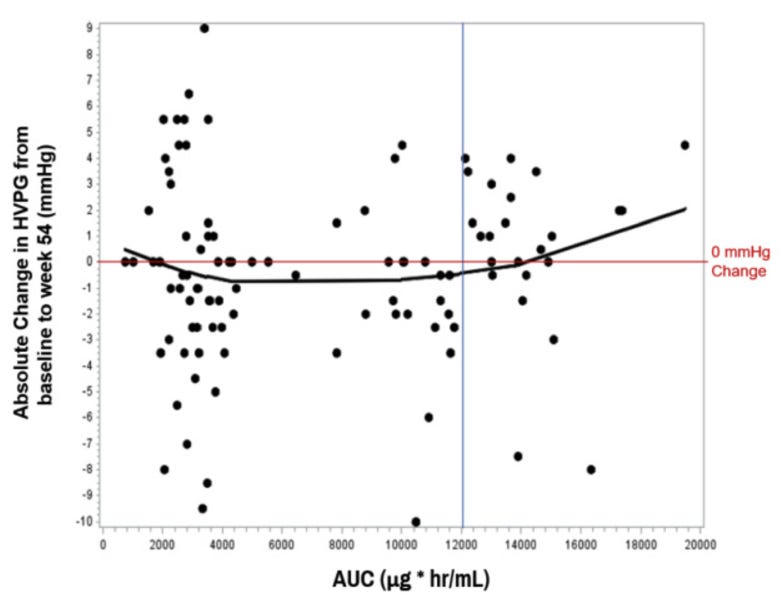
This appears to be management stretching here - you would conceptually expect a dose dependent effect with the Gal-3 mechanism, where more galectin inhibition improves inflammation and thus portal pressure.
Additionally, the subgroup analysis was conducted using a modified-ITT (excluding the 11 participants who discontinued out of 151 who completed the study). There was also no central reading of the endoscopic findings, which makes the rate of varices development less reliable.
Only an unplanned post-hoc analysis of the subgroup without varices was significant (p=0.02). However, because <80% of groups had <5 expected events in the chi^2 test - the company should actually have conducted a Fischer’s exact test vs. chi^2 shared.

If you run the numbers on the Fischer’s exact test and adjust the p-value for the subgroup analysis (p = 0.025 threshold), the p-value is 0.027 and is not significant. The Phase 3 never had any validation to be run.
3) Molecular Binding: Belapectin binds to the Gal-3 CRD & N-terminal domain to disrupt the fibrosis lattice (i.e., fibrosome). GALT management is pursuing belapectin as a first-in-class small molecule inhibitor of galectin-3, claiming a specific focus on its allosteric binding profile (vs. CRD-only).
While conceptually we acknowledge there is evidence suggesting non-specific, allosteric binding is possible (vs. predominantly CRD for most binding proteins to Gal-3), there is limited data to justify these nonspecific interactions (large, complex belapectin polymer binding to 5 galectin-3s) outside of GALT’s publications. To be clear - it’s 100% possible these effects do exist - we are merely observing the data shared. Ideally - the binding structure for belapectin & Gal-3 would be shared for allosteric sites - not just from the direct CRD binding.
Structure and binding
Belapectin is a galactoarabinan-rhamnogalacturonan composed of galacturonic acid, galactose, arabinose, rhamnose, and smaller amounts of other sugars. Belapectin is thought to bind to galectin-3.
Previous investigations frame Gal-3 by poor druggability due to 1) its large and diverse carbohydrate recognition domain (CRD) binding site that make it difficult for a lock and key small molecule mechanism; 2) the specific arrangement of Gal-3 recognizes primarily oxygen atoms, which is optimal for the recognition of β-galactoside hydroxyl residues & thus hard to outcompete with a drug; and 3) the rapid exchange of water molecules that may interfere with stable binding.
Management is pushing forward the thesis that belapectin also binds alternate allosteric sites on the Gal-3 molecule. Management shared a single experiment where belapectin bound to galectin-3 at a 1:5 ratio at both the CRD domain (S-face and F-face of C-terminus) and N-terminal domain (disrupts fibrosome construction). Management has affirmed this binding profile and previously shared a KD of 2.9 μM at Gal-3 and 8 μM at Gal-1.
While we do believe it’s conceptually possible that belapectin does indeed bind allosteric sites on the Gal-3 molecule, non-specific interactions from a single experiment at lesser known binding sites is not sufficient evidence to claim these binding events do truly occur.
At the very least, the PoS should be lower for studies relying on in vitro studies of ECM interactions - where in humans other molecules in the ECM will likely disrupt those non-specific interactions.
Figure 1: Structure and binding characteristics of belapectin

Figure 2: Belapectin mechanism of action
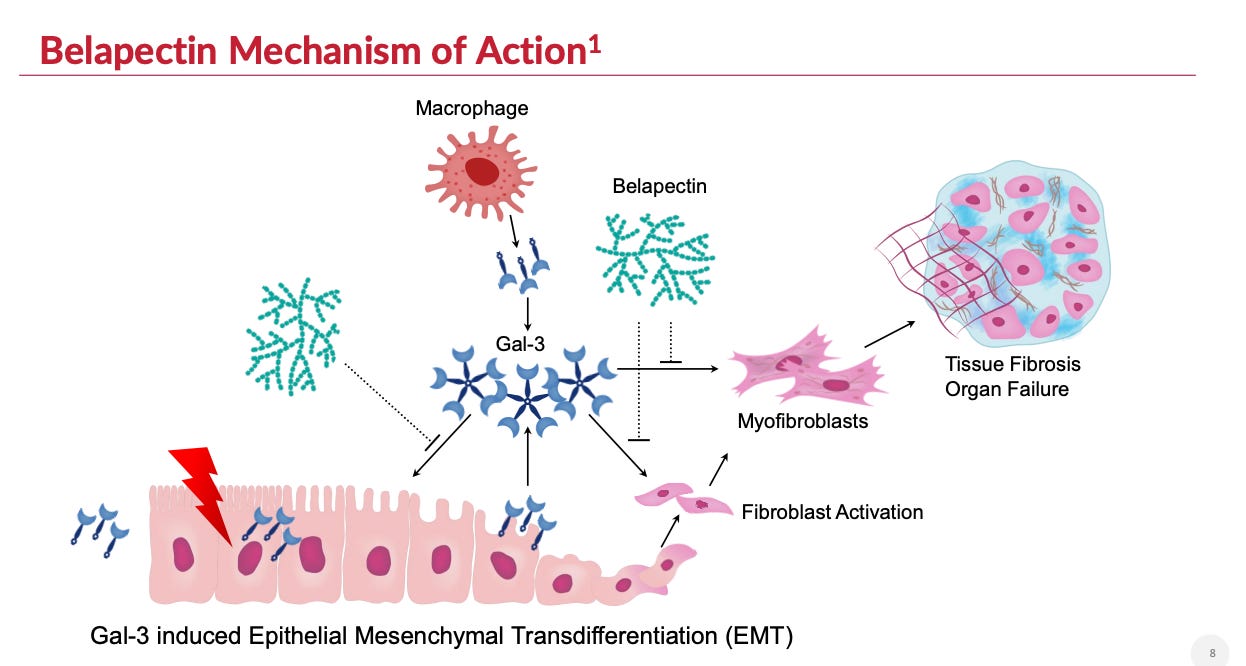
Mechanism of action
Galectin-3 inhibition impacts many pathways, but the most direct pathway on myofibroblast activation is likely through limiting TGF-beta receptor activation of myofibroblasts.
Resistance to blood flow is caused by cirrhosis, resulting in portal hypertension, retrograde flow, and finally congested veins in the portal system (i.e., varices). Portal myofibroblasts play a significant role in the development of portal hypertension and varices. Portal myofibroblasts primarily contribute to liver fibrosis by producing collagen and fibrosis (which belapectin targets). However, they also promote angiogenesis and have an inherent contractile state (from the smooth muscle, myo- in myofibroblasts) that can increase intrahepatic vascular resistance (BP = cardiac output x systemic vascular resistance). Therefore, you would feel good about the mechanism if the fibrosis AND smooth muscle actin reductions were clear in the preclinical/clinical data (which, as discussed above - they aren’t).
4) Short patent life
According to the patent, belapectin exclusivity will end in 2032 (management says 2035). Since the timelines for MASH trials are long, we wouldn’t expect approval until 2028-2029 at the very earliest - 3 years on market makes this a poor acquisition or commercialization target.
Studying peer trial designs gives us a clue into when the phase III will read out. MDGL phase 3 trial initiation to approval was 5 years. 89bio presented phase 2b 1Q23 and started phase 3 in 1Q/2Q24. GALT is presenting their phase 2b and would have to run the long MASH Phase 3 trials - likely case for approval is 4-5 years from now (~2028-2029).
5) High dilutive risk
Management is burning ~$45M/yr annualized ($9M Q324) with ~$25M cash on hand in an expensive indication (MASH Phase 3 trials cost ~$200M each). Realistic to expect >60% risk of dilution prior to PDUFA, suggesting a near-term significant raise that would put downward pressure on the stock even if the trial is successful.
6) Upcoming catalyst
NAVIGATE Data YE24: Primary readout for Phase 2b NAVIGATE trial, where management will report on the emergence of varices at 18 months.
The notable changes from the prior Phase 2b are: 1) Not requiring liver biopsies and 2) Only enrolling patients without varices on EGD. They are trying to get healthier patients by loosening the restrictions on portal hypertension as well - from requiring HVPG measurement >= 6 mm Hg (prior trial) to allowing reduced platelet counts or other clinical indicators to serve as evidence of portal hypertension. We could see not requiring liver biopsies going either way - you may get sicker patients since those are the ones who will have a more significant cirrhosis history and thus more likely to be enrolled.
Based on our analysis, for the upcoming readout - assuming placebo responds the same amount as the prior trial (17.6% of patients develop new varices, 21 patients vs. 18% prev. shown), our analysis demonstrates that 10 patients (8.4%) at the most may develop varices and still show statistical significance (off a chi^2 test at p=0.05 for the 2mg subgroup). Note you can argue a multiplicity adjustment is required here given the team will push the drug forward if EITHER the 2mg or 4mg subgroup show an effect (ie the hurdle is even higher).

They’ve already shared baseline criteria that supports our hypothesis that these are easier to treat patients.
Liver stiffness average in cirrhosis is 24.8kPA (range 5.5–75.4 usually for cirrhosis patients) so this is a healthier population
MELD score (6-40) is on the lower end - healthier population w compensated disease
Child pugh score - class A5 or well compensated disease
GLP-1s in 23% of patients
Valuation - share price at $2.85; cash is ~$0.50. Short interest is <15% at time of writing - which across the universe of SMID bio is at the lower end of the range.
